scGenePT Perturbation Prediction Tutorial
Last Updated: [12/04/2024] This notebook offers examples of how to use trained scGenePT models in inference mode for perturbation prediction. It uses models fine-tuned on the Norman dataset [1] and offers examples of predicting post-perturbation expression responses for single gene perturbations: POU3F2, CDKN1B and gene combinations: SAMD1+ZBTB1. Models have not been evaluated on multiple gene combination perturbation responses, so behavior is unknown.
Model : scGenePT is a collection of single-cell models for perturbation prediction. It leverages the
scGPT[2]model for scRNAseq data by injecting language embeddings at the gene level into the model architecture. The language gene embeddings are obtained by embedding gene level information from different knowledge sources using LLMs. The knowledge sources used include NCBI gene descriptions, UniProt protein Summaries for protein coding genes - as inspired by the genePT [3] approach - and GO (Gene Ontology) Gene Molecular Annotations, across three different axes: Molecular Function, Biological Process and Cellular Component. The model variations available are:
- scGenePT_NCBI = scGPT + NCBI Gene Card Summaries
- scGenePT_NCBI+UniProt = scGPT + NCBI Gene Card Summaries + UniProt Protein Summaries
- scGenePT_GO-F = scGPT + GO Molecular Functions Annotations
- scGenePT_GO-C = scGPT + GO Cellular Components Annotations
- scGenePT_GO-P = scGPT + GO Biological Processes Annotations
- scGenePT+GO-all = scGPT + GO_F + GO_C + GO_P
In this tutorial, we will focus on comparing scGenePT_GO-C and scGenePT_NCBI+UniProt models.
Dataset: The Norman Dataset [1] is a CRISPR perturb-seq dataset containing single and two-gene perturbations. We use a processed version of the dataset that contains 105 single and 131 two-gene combinations perturbations coming from 91k observations. Cells in the dataset are log-normalized and filtered to the top 5000 highly variable genes. Models in ths notebook are trained on the train split of the dataset. We offer examples of:
- performing inference on the test split of the Norman dataset
- performing inference on a random control sample and on a new anndata file
Hardware Requirements: We strongly recommend running this notebook on a GPU instance. It should run on a CPU as well, but it will be much slower.
The notebook is structured and offers the following examples:
- Perturbation prediction
-
Plotting the top 20 Differentially Expressed Genes post-perturbation
- Predicting perturbation response for perturbing the POU3F2 gene using a scGenePT_GO−C model
- Predicting perturbation response for perturbing the CDKN1B gene using a scGenePT_GO−C model
- Predicting perturbation response for perturbing the SAMD1+ZBTB1 gene combination using a scGenePT_NCBI+UniProt model
- Perturbation prediction on NumPy arrays holding control samples
- Perturbation prediction on AnnData files
Setup
We start by installing the required packages needed to run the notebook, as well as adding the needed data. Note that this section might take a few minutes to run:
%%capture [--no-stderr]
!pip -q install torch==2.1.2 torch_geometric==2.5.3 torchdata==0.7.1 torchmetrics==1.4.0.post0 packaging==24.1 numpy==1.26.4 scgpt awscli!git clone https://github.com/czi-ai/scGenePT.gitfrom pathlib import Path
def initialize_folder_structure(models_dir, models, dataset):
models_finetuned = models_dir / "finetuned"
model_embeddings = models_dir / "gene_embeddings"
model_embeddings_annotations = models_dir / "gene_embeddings" / "gene_annotations"
models_finetuned.mkdir(parents=True, exist_ok=True)
for model in models:
model_dir = models_finetuned / model / dataset
model_dir.mkdir(parents=True, exist_ok=True)
model_embeddings.mkdir(parents=True, exist_ok=True)
model_embeddings_annotations.mkdir(parents=True, exist_ok=True)# This creates the required folder structure needed for the data
initialize_folder_structure(Path('scGenePT/models'), ['scgpt', 'scgenept_go_c', 'scgenept_ncbi+uniprot'], "norman")Model and Data
The trained models can be downloaded from S3:
dataset_name = 'norman' # this can also be replaced with adamson, but the model files need to be updated accordingly throughout the notebook# Download the Gene Embeddings
!aws s3 cp --no-sign-request s3://czi-scgenept-public/models/gene_embeddings/GO_C_gene_embeddings-gpt3.5-ada-concat.pickle scGenePT/models/gene_embeddings/
!aws s3 cp --no-sign-request s3://czi-scgenept-public/models/gene_embeddings/NCBI+UniProt_embeddings-gpt3.5-ada.pkl scGenePT/models/gene_embeddings/
# Download the trained scGenePT models
!aws s3 sync --no-sign-request s3://czi-scgenept-public/models/finetuned/scgpt/{dataset_name}/ scGenePT/models/finetuned/scgpt/{dataset_name}/
!aws s3 sync --no-sign-request s3://czi-scgenept-public/models/finetuned/scgenept_go_c/norman/ scGenePT/models/finetuned/scgenept_go_c/{dataset_name}/
!aws s3 sync --no-sign-request s3://czi-scgenept-public/models/finetuned/scgenept_ncbi+uniprot/{dataset_name}/ scGenePT/models/finetuned/scgenept_ncbi+uniprot/{dataset_name}/Note that in this tutorial, we focus on comparing scGenePT_GO-C and scGenePT_NCBI+UniProt models, but all the other model variations can be downloaded in a similar way. The s3 links follow the structure:
s3://czi-scgenept-public/models/finetuned/{scgenept_model_name}/{dataset_name}So if you would like to download other model variations, you have to change the scgenept_model_name, and if you want to a model trained on a different dataset, you need to change the dataset_name.
For instance, to download a scGenePT_GO-P model trained on Adamson, you would run:
!aws s3 sync --no-sign-request s3://czi-scgenept-public/models/finetuned/scgenept_go_p/adamson/ scGenePT/models/finetuned/scgenept_go_p/adamson/Alternatively, you can mount all the assets at once - including all models and gene embeddings - by running the command below. Note that this will take a few min, as it will download ~10GB of data:
!aws s3 sync --no-sign-request s3://czi-scgenept-public/models scGenePT/modelsYour folder structure should now look something like:
- models
- pretrained
- scgpt/vocab.json
- gene_embeddings
- GO_C_gene_embeddings-gpt3.5-ada-concat.pickle
- NCBI+UniProt_embeddings-gpt3.5-ada.pkl
- gene_annotations
- gene_ontology_C.csv
- gene_ontology_P.csv
- gene_ontology_F.csv
- NCBI_summary_of_genes.json
- NCBI_UniProt_summary_of_genes.json
- finetuned
- scgpt/norman/best_model_rnd_seed_42_concat.pt
- scgenept_go_c/norman/best_model_gpt3.5_ada_rnd_seed_42_concat.pt
- scgenept_ncbi+uniprot/norman/best_model_gpt3.5_ada_rnd_seed_42_concat.pt
- tutorials
- scgenept_tutorial.ipynb%%capture [--no-stderr]
import sys
sys.path.insert(1, 'scGenePT')
from train import load_dataloader
from utils.data_loading import *
from models.scGenePT import *
import matplotlib.pyplot as plt
from gears.inference import evaluate, compute_metrics, deeper_analysis, non_dropout_analysis
import scanpy as sc
import pickle as pkl1. Perturbation Prediction using a trained scGenePT model
Load dataset
We start by inspecting the Norman dataset that the models were trained on. The dataloaders we used for training used a pre-processed version of Norman from GEARS [3]. Models in this notebook have been finetuned on the train split of this dataset. We can look at the AnnData file that contains all the data used for training/validation and testing. We will inspect the splits in a later section.
!aws s3 cp --no-sign-request s3://czi-scgenept-public/training_data/{dataset_name}_pert_data_adata.h5ad.gz scGenePT/tutorials/{dataset_name}_pert_data_adata.h5ad.gz
!gzip -d scGenePT/tutorials/{dataset_name}_pert_data_adata.h5ad.gzpert_adata = sc.read_h5ad(f'scGenePT/tutorials/{dataset_name}_pert_data_adata.h5ad')The code below can be used to get the same data directly from GEARS dataloaders. Note that it need ~20GB of memory and will time out on a T4 Google Collab instance. But it will run on an instance with larger RAM memory.
from gears import PertData
# Download the Pre-processed Norman dataset directly from GEARS
pert_data = PertData('./data')
pert_data = load_dataloader(dataset_name, batch_size, eval_batch_size, split = 'simulation')
train_loader = pert_data.dataloader['train_loader']
val_loader = pert_data.dataloader['val_loader']
test_loader = pert_data.dataloader['test_loader']
pert_adata = pert_data.adata
pert_data_subgroup = pert_data.subgroupDataset Exploration
This AnnData object contains gene expression counts and additional information about the cell observations:
pert_adata# Output
AnnData object with n_obs × n_vars = 91205 × 5045
obs: 'condition', 'cell_type', 'dose_val', 'control', 'condition_name'
var: 'gene_name'
uns: 'non_dropout_gene_idx', 'non_zeros_gene_idx', 'rank_genes_groups_cov_all', 'top_non_dropout_de_20', 'top_non_zero_de_20'
layers: 'counts'The gene expression counts are stored in pert_adata.X, where each entry (i, j) corresponds to the gene
expression counts of gene j in row i.
pert_adata.X # 902105 cells, 5045 genesThe pert_adata.obs file contains information about the different perturbation conditions applied to the data
stored in .X. For instance, below we can see examples of 1-gene perturbations: TSC22D1+ctrl, MAML2+ctrl, etc,
examples of 2-gene perturbations: KLF1+MAP2K6, CEBPE+RUNX1T1, and examples of control samples. Each of the conditions
corresponds to gene expression counts stored in .X.
pert_adata.obs # perturbation conditions applied to the 902105 cells# Output:
condition cell_type dose_val control condition_name
cell_barcode
AAACCTGAGGCATGTG-1 TSC22D1+ctrl A549 1+1 0 A549_TSC22D1+ctrl_1+1
AAACCTGAGGCCCTTG-1 KLF1+MAP2K6 A549 1+1 0 A549_KLF1+MAP2K6_1+1
AAACCTGCACGAAGCA-1 ctrl A549 1 1 A549_ctrl_1
AAACCTGCAGACGTAG-1 CEBPE+RUNX1T1 A549 1+1 0 A549_CEBPE+RUNX1T1_1+1
AAACCTGCAGCCTTGG-1 MAML2+ctrl A549 1+1 0 A549_MAML2+ctrl_1+1
... ... ... ... ... ...
TTTGTCAGTCATGCAT-8 RHOXF2BB+SET A549 1+1 0 A549_RHOXF2BB+SET_1+1
TTTGTCATCAGTACGT-8 FOXA3+ctrl A549 1+1 0 A549_FOXA3+ctrl_1+1
TTTGTCATCCACTCCA-8 CELF2+ctrl A549 1+1 0 A549_CELF2+ctrl_1+1
TTTGTCATCCCAACGG-8 BCORL1+ctrl A549 1+1 0 A549_BCORL1+ctrl_1+1
TTTGTCATCTGGCGAC-8 MAP4K3+ctrl A549 1+1 0 A549_MAP4K3+ctrl_1+1
91205 rows × 5 columnsThe pert_adata.var gives us the set of genes:
print(f'There are {len(pert_adata.var)} number of genes present in the dataset.')
pert_adata.var.head() # the names of the 5045 genes# Output:
There are 5045 number of genes present in the dataset.
gene_name
gene_id
ENSG00000239945 RP11-34P13.8
ENSG00000223764 RP11-54O7.3
ENSG00000187634 SAMD11
ENSG00000187642 PERM1
ENSG00000188290 HES4Load trained scGenePT models
In this section, we show examples of loading multiple scGenePT models for prediction in order to compare them. All the models under the scGenePT Model Zoo can be found under GitHub repo and the trained model weights under s3://czi-scgenept-public/models.
Here, we load the following models
- scGPT = scGPT
- scGenePT_NCBI+UniProt = scGPT + NCBI Gene Card Summaries + UniProt Protein Summaries
- scGenePT_GO-C = scGPT + GO Cellular Components Annotations
Note that models are assumed to be under the models/finetuned/{dataset_name}/{model_type}/ folder.
def load_trained_scgenept_model(adata, model_type, models_dir, model_location, device, verbose = False):
embs_to_include = get_embs_to_include(model_type)
vocab_file = models_dir + 'pretrained/scgpt/vocab.json'
vocab, gene_ids, dataset_genes, gene2idx = match_genes_to_scgpt_vocab_from_adata(vocab_file, adata, SPECIAL_TOKENS)
ntokens = len(vocab) # size of vocabulary
genept_embs, genept_emb_type, genept_emb_dim, found_genes_genept = initialize_genept_embeddings(embs_to_include, dataset_genes, vocab, model_type, models_dir)
go_embs_to_include, go_emb_type, go_emb_dim, found_genes_go = initialize_go_embeddings(embs_to_include, dataset_genes, vocab, model_type, models_dir)
# we disable flash attention for inference for simplicity
use_fast_transformer = False
model = scGenePT(
ntoken=ntokens,
d_model=EMBSIZE,
nhead=NHEAD,
d_hid=D_HID,
nlayers=NLAYERS,
nlayers_cls=N_LAYERS_CLS,
n_cls=N_CLS,
vocab=vocab,
n_perturbagens=2,
dropout=0.0,
pad_token=PAD_TOKEN,
pad_value=PAD_VALUE,
pert_pad_id=PERT_PAD_ID,
use_fast_transformer=use_fast_transformer,
embs_to_include = embs_to_include,
genept_embs = genept_embs,
genept_emb_type = genept_emb_type,
genept_emb_size = genept_emb_dim,
go_embs_to_include = go_embs_to_include,
go_emb_type = go_emb_type,
go_emb_size = go_emb_dim
)
pretrained_params = torch.load(model_location, weights_only=True, map_location = device)
if not use_fast_transformer:
pretrained_params = {
k.replace("Wqkv.", "in_proj_"): v for k, v in pretrained_params.items()
}
model.load_state_dict(pretrained_params)
if verbose:
print(model)
model.to(device)
return model, gene_idsdevice = torch.device("cuda" if torch.cuda.is_available() else "cpu")
print(f'Using device {device}')
# Mapping from model name to the name of the model file model weights are saved
model_name2model_variation = {'scgpt' : 'best_model_seed_42.pt',
'scgenept_ncbi+uniprot_gpt' : 'best_model_gpt3.5_ada_rnd_seed_42.pt',
'scgenept_go_c_gpt_concat' : 'best_model_gpt3.5_ada_rnd_seed_42_concat.pt'}
# Names of the scGenePT models to load. Note that these have to match the keys in the model_name2model_variation dict
models = ['scgpt', 'scgenept_go_c_gpt_concat', 'scgenept_ncbi+uniprot_gpt']
# To load all the models, uncomment this code and download the specific models from S3 using:
# !aws s3://czi-scgenept-public/models/finetuned/{scgenept_model_name}/{dataset_name} scGenePT/models/finetuned/{scgenept_model_name}/{dataset_name}
# model_name2model_variation = {'scgpt' : 'best_model_seed_42.pt',
# 'scgenept_ncbi_gpt' : 'best_model_gpt3.5_ada_rnd_seed_42.pt',
# 'scgenept_ncbi+uniprot_gpt' : 'best_model_gpt3.5_ada_rnd_seed_42.pt',
# 'scgenept_go_c_gpt_concat' : 'best_model_gpt3.5_ada_rnd_seed_42_concat.pt',
# 'scgenept_go_f_gpt_concat' : 'best_model_gpt3.5_ada_rnd_seed_42_concat.pt',
# 'scgenept_go_p_gpt_concat' : 'best_model_gpt3.5_ada_rnd_seed_42_concat.pt',
# 'scgenept_go_all_gpt_concat' : 'best_model_gpt3.5_ada_rnd_seed_42_concat.pt'}
# models = ['scgpt', 'scgenept_go_c_gpt_concat', 'scgenept_go_p_gpt_concat', 'scgenept_go_f_gpt_concat', 'scgenept_go_all_gpt_concat', 'scgenept_ncbi_gpt', 'scgenept_ncbi+uniprot_gpt']
trained_models = {}
# Location of where the pretrained scGPT model and gene embeddings are located
pretrained_scgpt_model_dir = 'scGenePT/models/'
for model_name in models:
print(f"Now loading a {model_name} model ... ")
print('=' * 30)
model_filename = model_name2model_variation[model_name]
if model_name != 'scgpt':
model_prefix = ''.join(model_name.split('_gpt')[:-1])
else:
model_prefix = model_name
model_location = f'scGenePT/models/finetuned/{model_prefix}/{dataset_name}/{model_filename}'
model, gene_ids = load_trained_scgenept_model(pert_adata, model_name, pretrained_scgpt_model_dir, model_location, device)
print('Done!\n')
trained_models[model_name] = model# Output:
Using device cuda
Now loading a scgpt model ...
==============================
scGenePT model-type: scgpt
match 4547/5045 genes in vocabulary of size 60697.
Using the following embeddings:['scGPT_counts_embs', 'scGPT_token_embs']
Done!
Now loading a scgenept_go_c_gpt_concat model ...
==============================
scGenePT model-type: scgenept_go_c_gpt_concat
match 4547/5045 genes in vocabulary of size 60697.
Using c GO embs
Matched 2945 out of 5045 genes in the GenePT-w embedding
Using the following embeddings:['GO_token_embs_gpt_concat', 'scGPT_counts_embs', 'scGPT_token_embs']
Done!
Now loading a scgenept_ncbi+uniprot_gpt model ...
==============================
scGenePT model-type: scgenept_ncbi+uniprot_gpt
match 4547/5045 genes in vocabulary of size 60697.
Using ncbi+uniprot genept embs, embedded with gpt
Matched 3351 out of 5045 genes in the GenePT-w embedding
Using the following embeddings:['scGPT_counts_embs', 'scGPT_token_embs', 'genePT_token_embs_gpt']
Done!2. Plot the Top Differentially Expressed Genes post-perturbation
We can use the trained models to perform inference. Below, we offer an example of how to do this on the test split of the Norman dataset - this is novel data the model has not been trained on. In the following sections we offer examples of performing inference on completely new data, either in numpy or anndata form.
We explore the following scenarios and showcase how to plot the top differentially expressed genes post-perturbation for:
- single-gene perturbation: predicting the effects of perturbing the POU3F2 gene and the CDKN1B gene individually. We offer examples of comparing scGPT with scGenePT_GO-C model predictions
- two-gene perturbation: predicting the effect of perturbing the SAMD1+ZBTB1 genes simultaneously. We offer an example of comparing scGPT and scGenePT_NCBI+UniProt model predictions
def plot_perturbation(
model: nn.Module, adata, query: str, model_type, color, marker, title, save_file: str = None, amp = True, pool_size: int = None
):
import seaborn as sns
import numpy as np
import matplotlib.pyplot as plt
sns.set_theme(style="ticks", rc={"axes.facecolor": (0, 0, 0, 0)}, font_scale=1.5)
adata_ctrl = adata[adata.obs['condition'] == 'ctrl']
gene_names = adata.var['gene_name'].to_list()
gene2idx = {gene: idx for idx, gene in enumerate(adata.var['gene_name'])}
cond2name = dict(adata.obs[["condition", "condition_name"]].values)
gene_raw2id = dict(zip(adata.var.index.values, adata.var.gene_name.values))
de_idx = [
gene2idx[gene_raw2id[i]]
for i in adata.uns["top_non_zero_de_20"][cond2name[query]]
]
genes = [
gene_raw2id[i] for i in adata.uns["top_non_zero_de_20"][cond2name[query]]
]
truth = adata[adata.obs.condition == query].X.toarray()[:, de_idx]
print(device)
pred = model.pred_perturb_from_ctrl(adata_ctrl, query, gene_names, device, gene_ids, amp, pool_size).squeeze()[de_idx]
ctrl_means = adata[adata.obs["condition"] == "ctrl"].to_df().mean()[de_idx].values
pred = pred - ctrl_means
truth = truth - ctrl_means
plt.figure(figsize=[16.5, 4.5])
plt.title(title + '\n' + query)
plt.boxplot(truth, showfliers=False, medianprops=dict(linewidth=0))
for i in range(pred.shape[0]):
_ = plt.scatter(i + 1, pred[i], color=color, marker=marker)
plt.axhline(0, linestyle="dashed", color="green")
ax = plt.gca()
ax.xaxis.set_ticklabels(genes, rotation=90)
plt.ylabel("Change in Gene Expression over Control", labelpad=10)
plt.tick_params(axis="x", which="major", pad=5)
plt.tick_params(axis="y", which="major", pad=5)
sns.despine()Before we perform predictions, we can explore the test split of the Norman dataset in more detail. We are interested in
this split, because this is data that the model has not seen during training, so performance on this data gives us a
sense of model performance on unseen data. The pert_data.subgroup object contains information about the set of
perturbations present in each of the train/val/test splits. These subgroups represent the perturbations in the AnnData
file pert_adata explored previously, that were used for training/validation or testing.
pert_data_subgroup = pkl.load(open(f'scGenePT/tutorials/{dataset_name}_pert_data_subgroups.pkl', 'rb'))
pert_data_subgroup.keys()The perturbation types in the test set are split into the following categories:
- unseen_single: single gene perturbations that have not been seen during training
- combo_seen0: two-gene perturbations, none of the genes has been seen perturbed (individually or as part of a combination) during training
- combo_seen1: two-gene perturbations, one of the genes has been seen perturbed (individually or as part of a combination) during training
- combo_seen2: two-gene perturbations, both genes has been seen perturbed (individually or as part of a combination) during training
unseen_single = pert_data_subgroup['test_subgroup']['unseen_single']
combo_seen0 = pert_data_subgroup['test_subgroup']['combo_seen0']
combo_seen1 = pert_data_subgroup['test_subgroup']['combo_seen1']
combo_seen2 = pert_data_subgroup['test_subgroup']['combo_seen2']
print(f'unseen_single: {len(unseen_single)}, combo_seen0: {len(combo_seen0)}, combo_seen1: {len(combo_seen1)}, combo_seen2: {len(combo_seen2)}')# Output:
unseen_single: 37, combo_seen0: 9, combo_seen1: 52, combo_seen2: 18Examples of combinations of genes where none of the genes has been seen during training:
combo_seen0# Output:
['POU3F2+FOXL2',
'ZBTB10+PTPN12',
'CEBPB+PTPN12',
'CBL+PTPN12',
'RHOXF2BB+SET',
'CDKN1C+CDKN1B',
'CDKN1C+CDKN1A',
'CDKN1B+CDKN1A',
'C3orf72+FOXL2']Single-Gene Perturbation: Predicting perturbation response for perturbing the POU3F2 gene
Then we can start exploring prediction on some of these perturbation categories. In the example below, we compare the
perturbation prediction responses of scGPT and scGenePT_GO-C - scGPT + GO Cellular Components Annotations by perturbing
the POU2F2 gene, which has not been seen perturbed during training by the model. We do this by sampling n = 300 random
controls from the training dataset and taking the mean of the average prediction for each control. According to
NCBI Gene Card, overexpression of the protein
encoded by POU3F2 is associated with an increase in the proliferation of melanoma cells. We can see that the genes
FABP5, HSP90AB1, PRDX1, NPM1, TMSB10, PTMA are all better predicted as having a negative fold change over control
by scGenePTGO-C, compared to scGPT which predicts a non-significant effect.
pert = 'POU3F2+ctrl'
colors = ['blue', 'fuchsia']
marker_types = [ 'o','s']
models_to_predict = ['scgpt', 'scgenept_go_c_gpt_concat']
for model, color, marker_type, title in zip(models_to_predict, colors, marker_types, ['scGPT', 'scGenePT_GO-C']):
plot_perturbation(trained_models[model], pert_adata, pert, model, color, marker_type, title, amp = True, pool_size=300)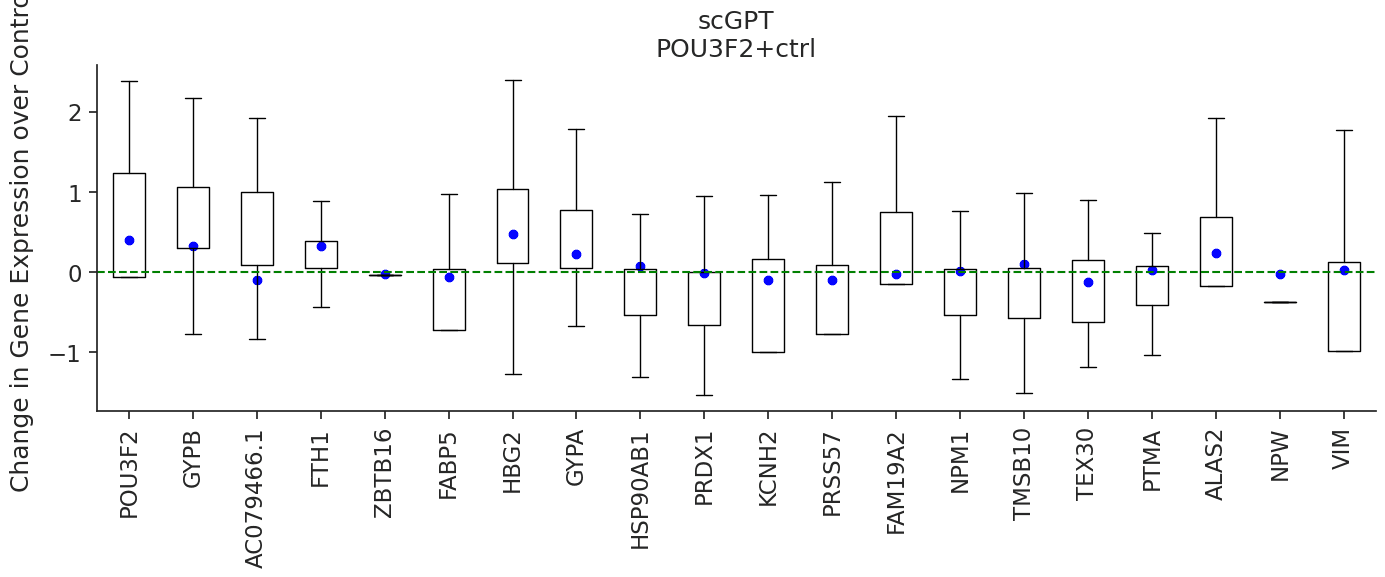
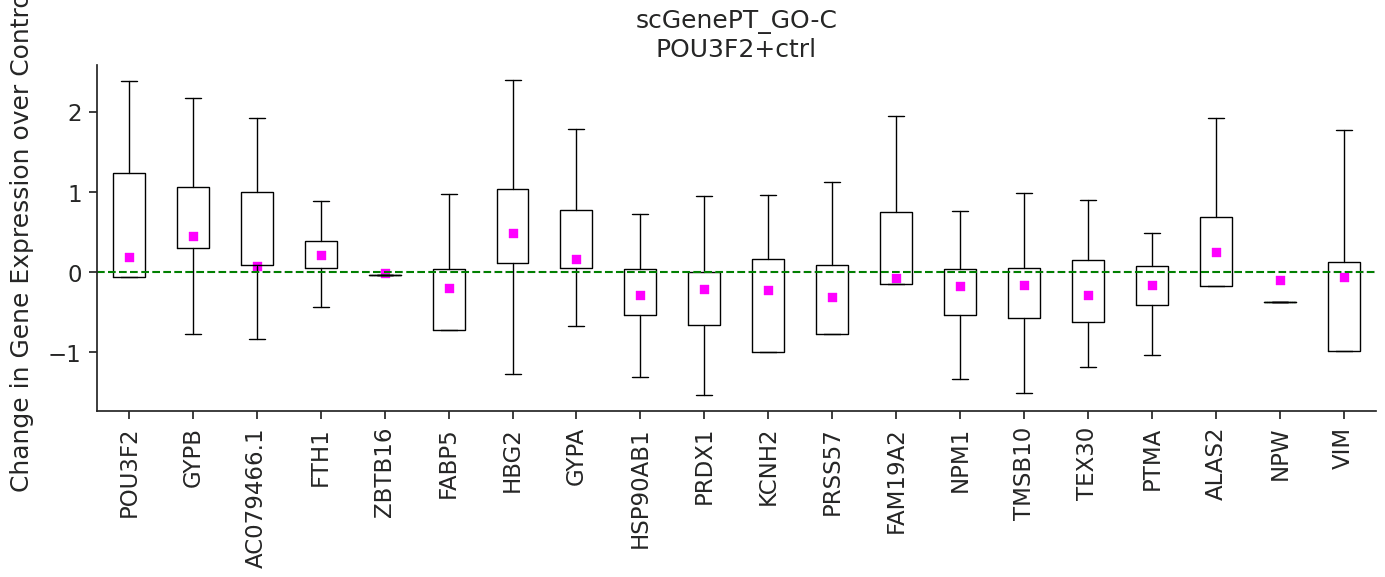
We can inspect the GO Gene Annotations that were used for this gene during training. If you don't already have the GO annotations downloaded, they can be retrieved from s3 through:
aws s3 sync --no-sign-request s3://czi-scgenept-public/models/gene_embeddings/gene_annotations/ scGenePT/models/gene_embeddings/gene_annotations/import pandas as pd
gene_annotations_dir = 'scGenePT/models/gene_embeddings/gene_annotations/'
#GO-C Cellular Component Annotations
GO_C_annotations_df = pd.read_csv(gene_annotations_dir + 'gene_ontology_C.csv')
#GO-P Biological Process Annotations
GO_P_annotations_df = pd.read_csv(gene_annotations_dir + 'gene_ontology_P.csv')
#GO-F Molecular Function Annotations
GO_F_annotations_df = pd.read_csv(gene_annotations_dir + 'gene_ontology_F.csv')For example, we can look at the GO-C Cellular Components annotations for the POU3F2 gene:
GO_C_annotations_df[GO_C_annotations_df['gene'] == 'POU3F2']# Output:
gene gene_name ontology_aspect direct_class_label
37738 POU3F2 POU domain, class 3, transcription factor 2 C nucleoplasm
37739 POU3F2 POU domain, class 3, transcription factor 2 C chromatin
37740 POU3F2 POU domain, class 3, transcription factor 2 C transcription regulator complexBy adding the GO Cellular Component annotations, the model learns that this gene is localized mostly in: nucleoplasm, chromatin and transcription regulator complex. Similarly, it learns about the subcellular localization of the other genes. Localization of gene products in the cell plays an important role in their biological function, e.g. protein-protein interaction; regulation of gene expression, transportation of protein. This tells us that subcellular localization is helpful in being able to predict effects of perturbation of this gene.
Single-Gene Perturbation: Predicting perturbation response for perturbing the CDKN1B gene
We can do this analysis for other genes as well. For example, the CDKN1B gene is another gene that has not been seen perturbed during training. According to this gene's NCBI Gene Card, mutations in this gene are associated with multiple enodcrine neoplasia type IV. We can see that scGenePTGO-C predicts HSP90AA1, PTMA, RANBP1, CKS1B, PRDX1, PHF19 and NME1 as correctly down-regulated, as opposed to scGPT which predicts either neutral effect or positive fold change. Similarly, we speculate that the model learns to incorporate cellular location information to better predict gene expression change in response to genetic perturbation.
pert = 'CDKN1B+ctrl'
colors = ['blue', 'red']
marker_types = [ 'o','s']
models_to_predict = ['scgpt', 'scgenept_go_c_gpt_concat']
for model, color, marker_type, title in zip(models_to_predict, colors, marker_types, ['scGPT', 'scGenePT_GO-C']):
plot_perturbation(trained_models[model], pert_adata, pert, model, color, marker_type, title, amp = True, pool_size=300)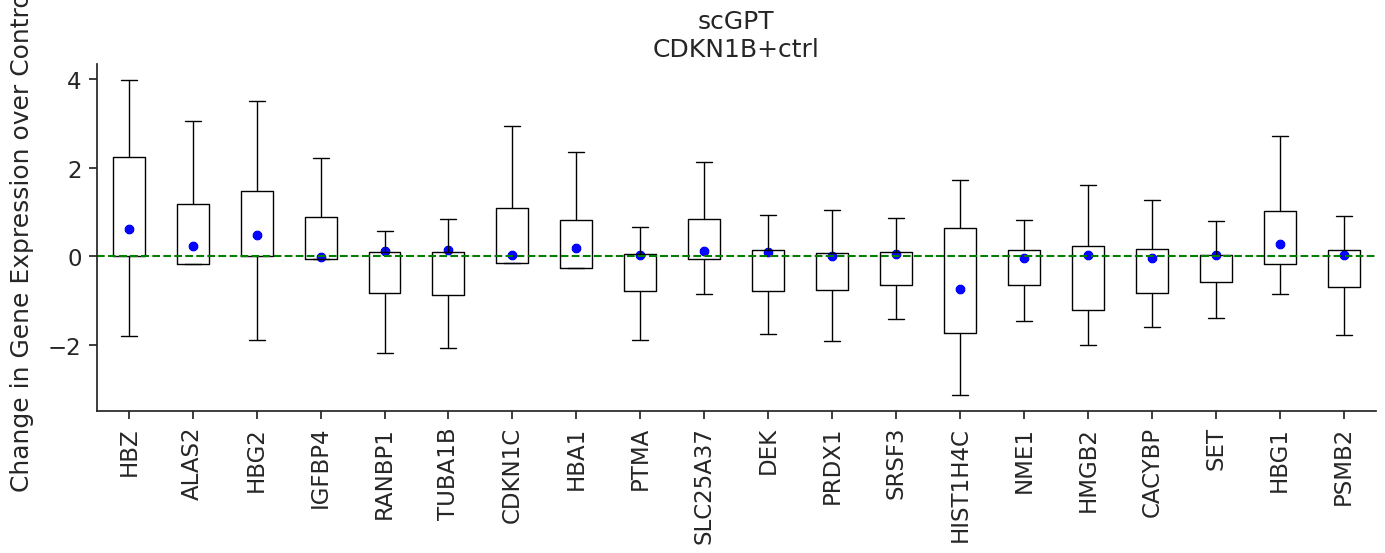
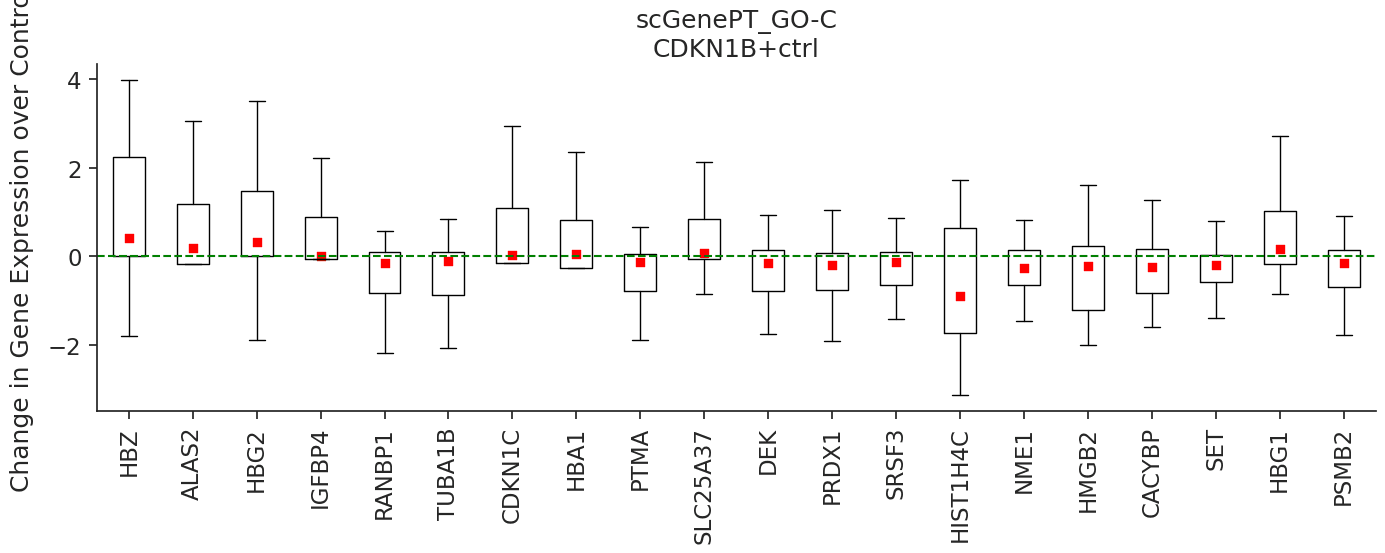
We can see that the subcellular localization annotations that the model sees during training for the CDK1NB gene are:
GO_C_annotations_df[GO_C_annotations_df['gene'] == 'CDKN1B']# Output:
gene gene_name ontology_aspect direct_class_label
72395 CDKN1B Cyclin-dependent kinase inhibitor 1B C nucleus
72396 CDKN1B Cyclin-dependent kinase inhibitor 1B C nucleoplasm
72397 CDKN1B Cyclin-dependent kinase inhibitor 1B C cytoplasm
72398 CDKN1B Cyclin-dependent kinase inhibitor 1B C endosome
72399 CDKN1B Cyclin-dependent kinase inhibitor 1B C cytosol
72400 CDKN1B Cyclin-dependent kinase inhibitor 1B C intracellular membrane-bounded organelle
72401 CDKN1B Cyclin-dependent kinase inhibitor 1B C Cul4A-RING E3 ubiquitin ligase complexWe speculate that having access to this information during training is helping the model make better predictions on the effect of perturbing this gene.
Two-gene Perturbation: Predicting perturbation response for perturbing the gene combination FOXA1+FOXL2
We can also look at effects of combination of genes. Below, we offer an example of predicting perturbation responses for the FOXA1+FOXL2 gene combination.
Note that you can experiment with different gene combinations in combo_seen0, combo_seen1, or combo_seen2
pert = 'FOXA1+FOXL2'
colors = ['blue', 'green']
marker_types = [ 'o', 's', 's']
models_to_predict = ['scgpt', 'scgenept_ncbi+uniprot_gpt']
for model, color, marker_type, title in zip(models_to_predict, colors, marker_types, ['scGPT', 'scGenePT_NCBI+UniProt']):
plot_perturbation(trained_models[model], pert_adata, pert, model, color, marker_type, title, amp = True, pool_size=300)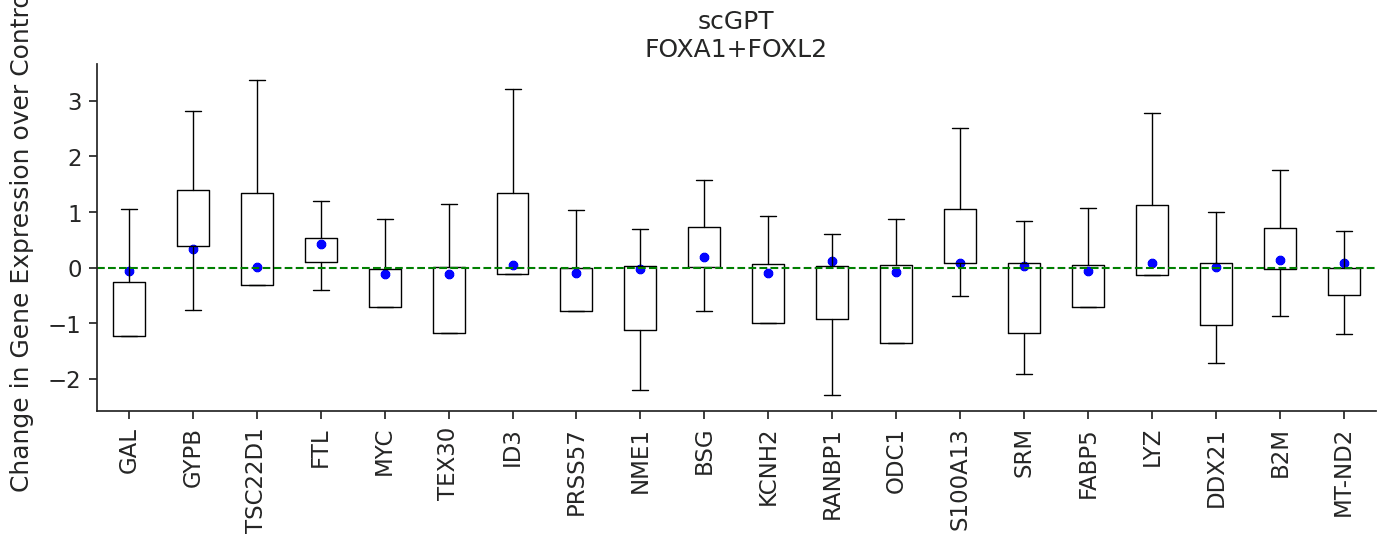
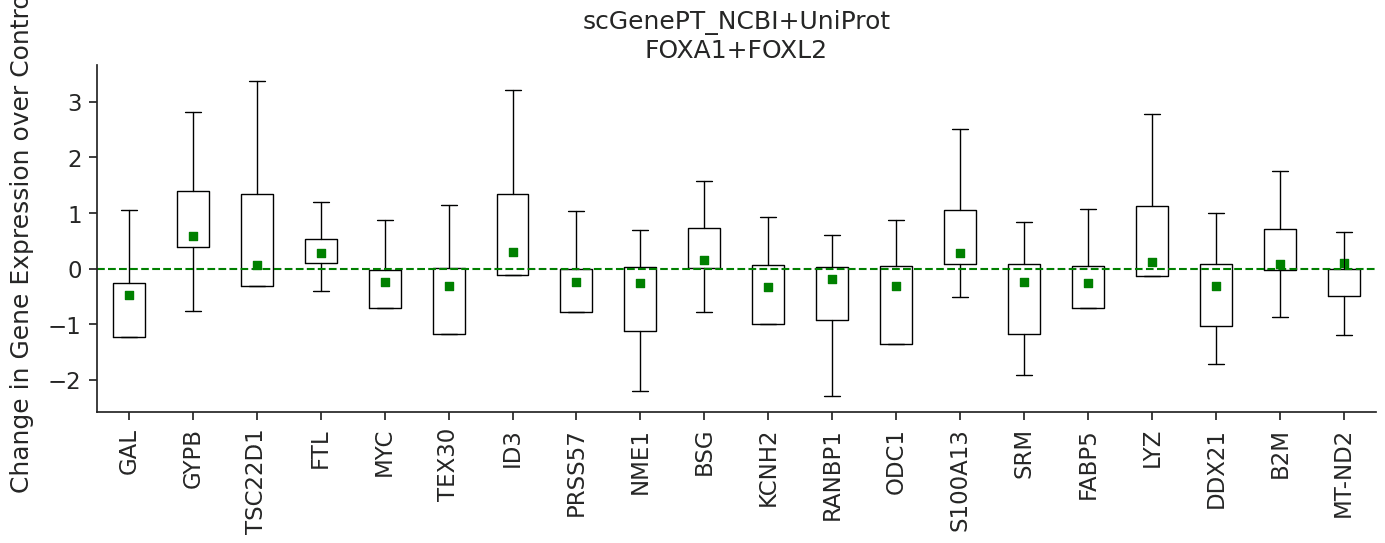
Similarly, we can look at the NCBI and UniProt annotations for the genes:
# NCBI Gene Card Annotations
NCBI_gene_card_summaries = json.load(open(gene_annotations_dir + 'NCBI_summary_of_genes.json', 'rb'))
# NCBI Gene Card + UniProt protein summaries Annotations
NCBI_UniProt_gene_card_protein_summaries = json.load(open(gene_annotations_dir + 'NCBI_UniProt_summary_of_genes.json', 'rb'))
comb_genes = pert.split('+')
for gene in comb_genes:
print(f"Annotations for GENE: {gene}")
print('='*30)
print(f"NCBI Gene Card Summary: {NCBI_gene_card_summaries[gene]}")
print('\n')
print(f"NCBI Gene Card + UniProt Protein Summary: {NCBI_UniProt_gene_card_protein_summaries[gene]}")
print('\n')# Output:
Annotations for GENE: FOXA1
==============================
NCBI Gene Card Summary: Gene Symbol FOXA1 This gene encodes a member of the forkhead class of DNA-binding proteins. These hepatocyte nuclear factors are transcriptional activators for liver-specific transcripts such as albumin and transthyretin, and they also interact with chromatin. Similar family members in mice have roles in the regulation of metabolism and in the differentiation of the pancreas and liver.
NCBI Gene Card + UniProt Protein Summary: Gene Symbol FOXA1 This gene encodes a member of the forkhead class of DNA-binding proteins. These hepatocyte nuclear factors are transcriptional activators for liver-specific transcripts such as albumin and transthyretin, and they also interact with chromatin. Similar family members in mice have roles in the regulation of metabolism and in the differentiation of the pancreas and liver. Protein summary: Transcription factor that is involved in embryonic development, establishment of tissue-specific gene expression and regulation of gene expression in differentiated tissues. Is thought to act as a 'pioneer' factor opening the compacted chromatin for other proteins through interactions with nucleosomal core histones and thereby replacing linker histones at target enhancer and/or promoter sites. Binds DNA with the consensus sequence 5'-[AC]A[AT]T[AG]TT[GT][AG][CT]T[CT]-3' (By similarity). Proposed to play a role in translating the epigenetic signatures into cell type-specific enhancer-driven transcriptional programs. Its differential recruitment to chromatin is dependent on distribution of histone H3 methylated at 'Lys-5' (H3K4me2) in estrogen-regulated genes. Involved in the development of multiple endoderm-derived organ systems such as liver, pancreas, lung and prostate; FOXA1 and FOXA2 seem to have at least in part redundant roles (By similarity). Modulates the transcriptional activity of nuclear hormone receptors. Is involved in ESR1-mediated transcription; required for ESR1 binding to the NKX2-1 promoter in breast cancer cells; binds to the RPRM promoter and is required for the estrogen-induced repression of RPRM. Involved in regulation of apoptosis by inhibiting the expression of BCL2. Involved in cell cycle regulation by activating expression of CDKN1B, alone or in conjunction with BRCA1. Originally described as a transcription activator for a number of liver genes such as AFP, albumin, tyrosine aminotransferase, PEPCK, etc. Interacts with the cis-acting regulatory regions of these genes. Involved in glucose homeostasis
Annotations for GENE: FOXL2
==============================
NCBI Gene Card Summary: Gene Symbol FOXL2 This gene encodes a forkhead transcription factor. The protein contains a fork-head DNA-binding domain and may play a role in ovarian development and function. Expansion of a polyalanine repeat region and other mutations in this gene are a cause of blepharophimosis syndrome and premature ovarian failure 3.
NCBI Gene Card + UniProt Protein Summary: Gene Symbol FOXL2 This gene encodes a forkhead transcription factor. The protein contains a fork-head DNA-binding domain and may play a role in ovarian development and function. Expansion of a polyalanine repeat region and other mutations in this gene are a cause of blepharophimosis syndrome and premature ovarian failure 3. Protein summary: Transcriptional regulator. Critical factor essential for ovary differentiation and maintenance, and repression of the genetic program for somatic testis determination. Prevents trans-differentiation of ovary to testis through transcriptional repression of the Sertoli cell-promoting gene SOX9 (By similarity). Has apoptotic activity in ovarian cells. Suppresses ESR1-mediated transcription of PTGS2/COX2 stimulated by tamoxifen (By similarity). Is a regulator of CYP19 expression (By similarity). Participates in SMAD3-dependent transcription of FST via the intronic SMAD-binding element (By similarity). Is a transcriptional repressor of STAR. Activates SIRT1 transcription under cellular stress conditions. Activates transcription of OSR23. Predicting on NumPy arrays holding control samples
We can make predictions on various data formats. Let's say we have a control sample held in a NumPy array. We can use one of the trained models to make predictions. Similarly as before, first we need to load the trained model:
model_name = 'scgenept_go_c_gpt_concat'
dataset_name = 'norman'
pert_adata = sc.read_h5ad(f'scGenePT/tutorials/{dataset_name}_pert_data_adata.h5ad')
model_filename = model_name2model_variation[model_name]
model_location = f'scGenePT/models/finetuned/scgenept_go_c/{dataset_name}/{model_filename}'
model, gene_ids = load_trained_scgenept_model(pert_adata, model_name, pretrained_scgpt_model_dir, model_location, device, verbose = False)# Output:
scGenePT model-type: scgenept_go_c_gpt_concat
match 4547/5045 genes in vocabulary of size 60697.
Using c GO embs
Matched 2945 out of 5045 genes in the GenePT-w embedding
Using the following embeddings:['GO_token_embs_gpt_concat', 'scGPT_counts_embs', 'scGPT_token_embs']gene_names = pert_adata.var['gene_name'].to_list()
print(f'There are {len(gene_names)} genes, the first 10 are: {gene_names[:10]}')# Output:
There are 5045 genes, the first 10 are: ['RP11-34P13.8', 'RP11-54O7.3', 'SAMD11', 'PERM1', 'HES4', 'ISG15', 'RP11-54O7.18', 'RNF223', 'LINC01342', 'TTLL10-AS1']Let's say we want to predict the effect of perturbing the FOSB gene.
# this can be replaced with another gene
pert = "FOSB+ctrl"
# the order of the genes in ctrl_sample also has to match that of gene_names
ctrl_sample = np.random.rand(5045)Then we need to assign the correct perturbation flags in the control sequence, so that the model knows what gene should get perturbed
def get_pert_flags(ctrl_sample, gene_names, pert):
pert_flags = np.zeros(len(ctrl_sample))
if pert!= 'ctrl':
for x in pert.split('+'):
if x != 'ctrl':
pert_flags[gene_names.index(x)] = 1
pert_flags = torch.from_numpy(pert_flags).long().to(device).unsqueeze(0)
return pert_flags
pert_flags = get_pert_flags(ctrl_sample, gene_names, pert)Now we can get the model predictions by calling the model in inference mode:
def pred_ctrl_sample(model, gene_ids, ctrl_sample, pert_flags, device):
ctrl_sample = torch.from_numpy(np.expand_dims(ctrl_sample, 0)).to(dtype = torch.float32).to(device)
gene_ids_tensor = torch.tensor(gene_ids).long().unsqueeze(0).to(device)
src_key_padding_mask = torch.zeros_like(gene_ids_tensor, dtype=torch.bool, device=device)
model = model.to(torch.float32)
with torch.cuda.amp.autocast(enabled=True):
with torch.no_grad():
output_dict = model(
gene_ids_tensor,
ctrl_sample,
pert_flags,
src_key_padding_mask=src_key_padding_mask,
CLS=False,
CCE=False,
MVC=False,
ECS=False,
do_sample=True,
)
prediction = output_dict["mlm_output"].float().detach().cpu().numpy()
return predictionpreds = pred_ctrl_sample(model, gene_ids, ctrl_sample, pert_flags, device)preds# Output:
array([[-2.4585724e-03, 3.7207031e-01, 1.1413574e-02, ...,
3.8906250e+00, 3.4814453e-01, 6.9946289e-02]], dtype=float32)preds.shape# Output:
(1, 5045)4. Predicting on AnnData files
We can follow a similar sequence to predict on an AnnData file. In the example below, you can replace the adata_ctrl
file with your file of choice:
adata_ctrl = pert_adata[pert_adata.obs['condition'] == 'ctrl'][:100] # this can be any other AnnData file
adata_ctrl# Output:
View of AnnData object with n_obs × n_vars = 100 × 5045
obs: 'condition', 'cell_type', 'dose_val', 'control', 'condition_name'
var: 'gene_name', 'id_in_vocab'
uns: 'non_dropout_gene_idx', 'non_zeros_gene_idx', 'rank_genes_groups_cov_all', 'top_non_dropout_de_20', 'top_non_zero_de_20'
layers: 'counts'The set and order of the genes in the AnnData file has to match that of the genes the model has been trained on. In most cases, this will mean filtering the data to match this. The 5045 genes used by the model are:
adata_ctrl.var# Output:
gene_name id_in_vocab
gene_id
ENSG00000239945 RP11-34P13.8 1
ENSG00000223764 RP11-54O7.3 -1
ENSG00000187634 SAMD11 1
ENSG00000187642 PERM1 1
ENSG00000188290 HES4 1
... ... ...
ENSG00000198786 MT-ND5 1
ENSG00000198695 MT-ND6 1
ENSG00000198727 MT-CYB 1
ENSG00000273554 AC136616.1 -1
ENSG00000278633 AC023491.2 -1
5045 rows × 2 columnsEach of them will correspond to values in the columns of adata_ctrl.X:
adata_ctrl.X # an entry (i, j) corresponds to the value of gene j in cell i; the 5045 genes must match to the list above# Output:
<100x5045 sparse matrix of type '<class 'numpy.float32'>'
with 40918 stored elements in Compressed Sparse Row format>To get a prediction for all the cells in adata_ctrl:
gene_pert = 'CEBPB+ctrl'
ctrl_size = None # Note that if ctrl_size = None, all ctrl samples are used; if ctrl_size != None, then ctrl_size samples will get randomly sampled.
return_mean = False # If this is True, then the mean of the predictions will get returned
preds = model.pred_perturb_from_ctrl(adata_ctrl, gene_pert, gene_names, device, gene_ids, pool_size = ctrl_size, return_mean = False).squeeze()preds# Output:
array([[-3.2157898e-03, 1.1054993e-02, 8.6441040e-03, ...,
3.8398438e+00, 1.1054993e-02, 1.1054993e-02],
[-3.3149719e-03, 1.0871887e-02, 8.5983276e-03, ...,
3.8378906e+00, 1.0871887e-02, 1.0871887e-02],
[-3.2272339e-03, 1.1085510e-02, 8.5754395e-03, ...,
3.8476562e+00, 1.1085510e-02, 1.1085510e-02],
...,
[-3.2882690e-03, 1.0841370e-02, 1.3641357e-02, ...,
3.8457031e+00, 1.0841370e-02, 1.0841370e-02],
[-3.2958984e-03, 1.0810852e-02, 8.4457397e-03, ...,
3.8359375e+00, 1.0810852e-02, 1.0810852e-02],
[-3.2043457e-03, 1.0848999e-02, 8.7585449e-03, ...,
3.8515625e+00, 1.0848999e-02, 1.0848999e-02]], dtype=float32)preds.shape# Output:
(100, 5045)We can then assign the predictions to the anndata file:
adata_ctrl.layers[f'{model_name}_predictions'] = predsadata_ctrl# Output:
AnnData object with n_obs × n_vars = 100 × 5045
obs: 'condition', 'cell_type', 'dose_val', 'control', 'condition_name'
var: 'gene_name', 'id_in_vocab'
uns: 'non_dropout_gene_idx', 'non_zeros_gene_idx', 'rank_genes_groups_cov_all', 'top_non_dropout_de_20', 'top_non_zero_de_20'
layers: 'counts', 'scgenept_go_c_gpt_concat_predictions'To get a mean prediction over ctrl_size control samples from adata_ctrl, we can run:
ctrl_size = 300
preds_mean = model.pred_perturb_from_ctrl(adata_ctrl, gene_pert, gene_names, device, gene_ids, pool_size = ctrl_size, return_mean = True).squeeze()preds_mean# Output:
array([-3.2865589e-03, 1.7323304e-02, 8.5574342e-03, ...,
3.8426368e+00, 1.0909805e-02, 1.0909805e-02], dtype=float32)preds_mean.shape# Output:
(5045,)Contact & Feedback
Ana-Maria Istrate, aistrate@chanzuckerberg.com
References
- Norman, Thomas M., et al. "Exploring genetic interaction manifolds constructed from rich single-cell phenotypes." Science 365.6455 (2019): 786-793.
- Cui, Haotian, et al. "scGPT: toward building a foundation model for single-cell multi-omics using generative AI." Nature Methods (2024): 1-11.
- Chen, Yiqun, and James Zou. "GenePT: a simple but effective foundation model for genes and cells built from ChatGPT." bioRxiv (2024): 2023-10.
- Roohani, Yusuf, Kexin Huang, and Jure Leskovec. "Predicting transcriptional outcomes of novel multigene perturbations with GEARS." Nature Biotechnology 42.6 (2024): 927-935.
Responsible Use
We are committed to advancing the responsible development and use of artificial intelligence. Please follow our Acceptable Use Policy when engaging with our services.
Should you have any security or privacy issues or questions related to the services, please reach out to our team at security@chanzuckerberg.com or privacy@chanzuckerberg.com respectively.